数据分析实践¶
分析所用数据目录结构
├── Sample_1
│ ├── 00_raw
│ │ └── *.fastq.gz
│ ├── 01_trimmed
│ ├── 02_assembly
│ ├── 03_clean
│ ├── 10_fasta
...
└── Sample_N
├── 00_raw
...
示例所使用数据
这里用一次 Miseq 测序一个 Run 的数据进行数据质控示例。本次 Run 一共对20个样品进行了 PE300 的测序。产生的测序数据所见如下:
$ ls
RN01_S01_L001_R1_001.fastq.gz RN06_S06_L001_R1_001.fastq.gz RN11_S11_L001_R1_001.fastq.gz RN16_S16_L001_R1_001.fastq.gz
RN01_S01_L001_R2_001.fastq.gz RN06_S06_L001_R2_001.fastq.gz RN11_S11_L001_R2_001.fastq.gz RN16_S16_L001_R2_001.fastq.gz
RN02_S02_L001_R1_001.fastq.gz RN07_S07_L001_R1_001.fastq.gz RN12_S12_L001_R1_001.fastq.gz RN17_S17_L001_R1_001.fastq.gz
RN02_S02_L001_R2_001.fastq.gz RN07_S07_L001_R2_001.fastq.gz RN12_S12_L001_R2_001.fastq.gz RN17_S17_L001_R2_001.fastq.gz
RN03_S03_L001_R1_001.fastq.gz RN08_S08_L001_R1_001.fastq.gz RN13_S13_L001_R1_001.fastq.gz RN18_S18_L001_R1_001.fastq.gz
RN03_S03_L001_R2_001.fastq.gz RN08_S08_L001_R2_001.fastq.gz RN13_S13_L001_R2_001.fastq.gz RN18_S18_L001_R2_001.fastq.gz
RN04_S04_L001_R1_001.fastq.gz RN09_S09_L001_R1_001.fastq.gz RN14_S14_L001_R1_001.fastq.gz RN19_S19_L001_R1_001.fastq.gz
RN04_S04_L001_R2_001.fastq.gz RN09_S09_L001_R2_001.fastq.gz RN14_S14_L001_R2_001.fastq.gz RN19_S19_L001_R2_001.fastq.gz
RN05_S05_L001_R1_001.fastq.gz RN10_S10_L001_R1_001.fastq.gz RN15_S15_L001_R1_001.fastq.gz RN20_S20_L001_R1_001.fastq.gz
RN05_S06_L001_R2_001.fastq.gz RN10_S10_L001_R2_001.fastq.gz RN15_S15_L001_R2_001.fastq.gz RN20_S20_L001_R2_001.fastq.gz
根据样品建立文件夹
首先我们对每一个样品的数据单独建立文件夹,并对 fastq 数据进行基本的质控。
# 新建以样本名为文件夹名,并移动数据到文件夹的 00_raw 目录下
$ for i in $(awk -F'L001' '{gsub("_$", "", $1); print $1}' < (ls *.fastq.gz) | sort | uniq ); \
> do mkdir -p $(basename $i) && mkdir -p $(basename $i)/00_raw; \
> mv $i*.fastq.gz $(basename $i)/00_raw/ ; done
# 查看建立的所有文件夹
$ ls -d
# 使用 FastQC 对测序数据进行质控分析
$ for i in $(ls -d RN*/00_raw/*.fastq.gz); do fastqc $i --extract -t 40 -q -o qc; done
$ python -m SimpleHTTPServer
打开浏览器,访问URL地址为服务器IP:8000,点击qc链接,里面有 fastqc 生成的各测序数据的 html 质控文件。我们以第一个样本 RN01_S01 为例,发现数据有以下问题:
- 250循环之后的数据质量不高(见图1)
- 序列有接头污染(见图2)
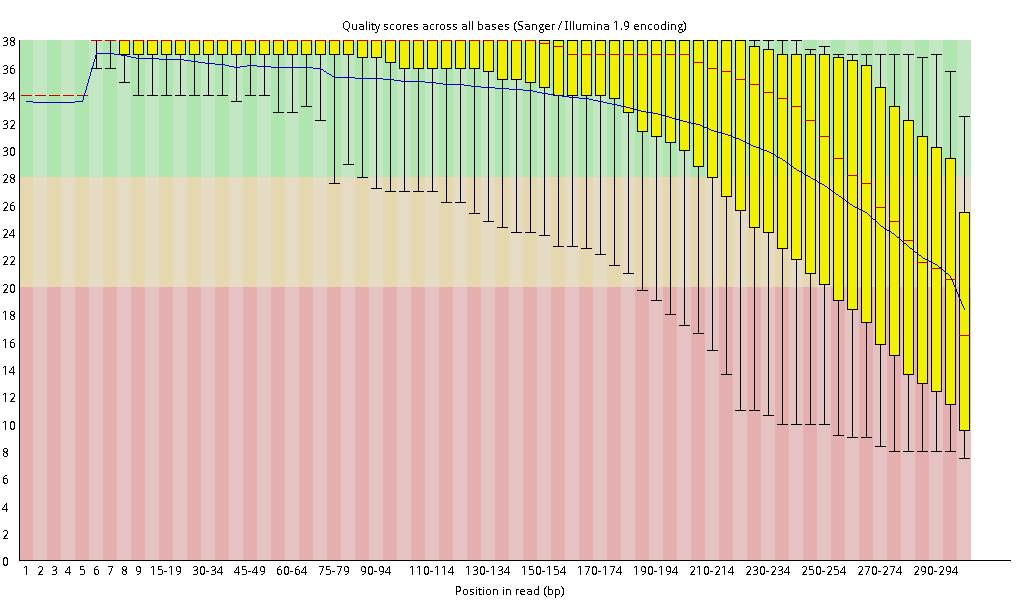 图1. 长读长区质量不高
图1. 长读长区质量不高
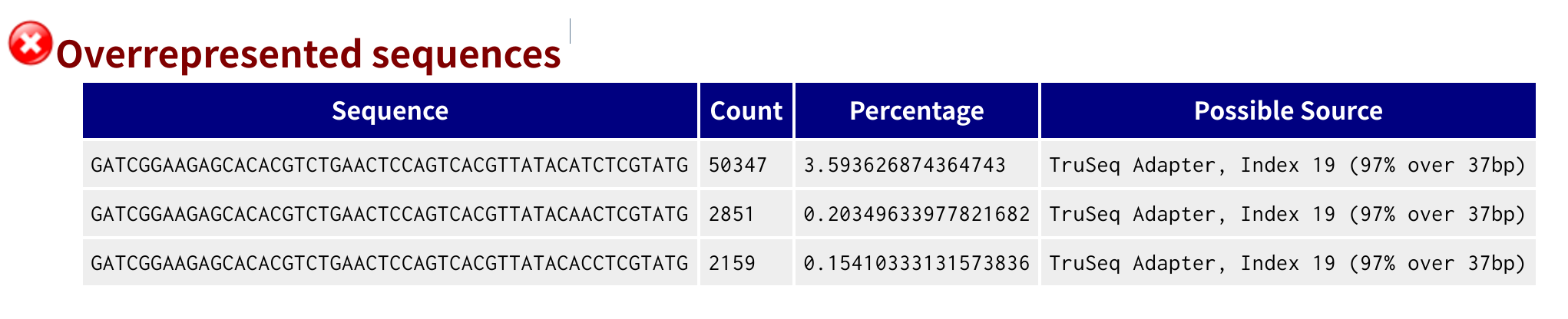 图2. 接头污染
图2. 接头污染
去除接头污染
首先我们要将污染的接头去除,采用 Trimmomatic 进行去除,同时也过滤低质量 reads 序列。
from fadapa import Fadapa
from Bio import SeqIO
from Bio.Seq import Seq
from Bio.SeqRecord import SeqRecord
from Bio.Alphabet import generic_nucleotide
import argparse
parser = argparse.ArgumentParser()
parser.add_argument('-i', action=store, dest='qc_file', type=str \
help='input your fastqc data')
args = parser.parse_args()
qc_file = Fadapa(args.qc_file)
if qc_file.summary()[-3][0] == 'fail':
adaptors = []
ors = qc_file.clean_data("Overrepresented sequences")
ors.pop(0)
for (index, seq) in enumerate(ors):
adaptors.append(SeqRecord(Seq(seq[0], generic_nucleotide), id="adaptor_%d" % (index+1), description=""))
SeqIO.write(adaptors, "adaptor.fasta", "fasta")
print "Overrepresented sequences has been save to adaptors.fasta"
else:
print "No Overrepresented sequences"
上面的 python 脚本使用 Biopython 和 Fadapa 模块将 FastQC 生成的过表达序列保存正接头文件,让 Trimmomatic 进一步处理。
$ echo 'alias trimm="java -jar /opt/Trimmomatic-0.36/trimmomatic-0.36.jar"' >> ~/.bashrc
$ source ~/.bashrc
$ trimm PE -threads 40 -phred33 \
> 00_raw/RN01_S01_L001_R1_001.fastq.gz 00_raw/RN01_S01_L001_R2_001.fastq.gz \
> 01_trim/R1_trimmed.fastq.gz 01_trim/R1_unpaired.fastq.gz \
> 02_trim/R2_trimmed.fastq.gz 01_trim/R2_unpaired.fastq.gz \
> ILLUMINACLIP:01_trim/adaptors/adaptor.fasta:2:30:10 \
> LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:50
初步拼接
先用 spades 进行拼接,了解基因组情况。
$ cd RN01_S01/00_raw/
$ spades.py -k 127 -t 40 --careful -1 01_trim/R1_trimmed.fastq.gz -2 01_trim/R2_trimmed.fastq.gz -o assembly/spades
$ du -h assembly/spades/scaffolds.fasta
10.1M assembly/spades/scaffolds.fasta
$ cat assembly/spades/scaffolds.fasta | grep '>' | tail -1
>NODE_3360_....
$ cat assembly/spades/scaffolds.fasta | grep '>' | awk -F'_' '{if ($6<10) print $0}' | wc -l
3104
结果获得的 scaffolds.fasta 文件大小为10M左右,而我们测序的目的物种基因组大小仅为3M;里面的 contigs 数量达到3360个,并且 contigs 平均覆盖度小于10的有3100多个,说明原始的测序数据很可能被其他物种污染了。这种污染可能发生在核酸提取,文库制备或者测序中(清洗管路不彻底)
观察覆盖度与污染序列的关系
用 Blast 的方法来筛选污染序列
# 下载污染物种的基因组数据,进行序列比对,看组装的nodes里那些是来源于污染物种。如果有多个污染物种,则可以将基因组数据合并 `cat 1.fa 2.fa 3.fa > containment.fasta`
$ makeblastdb -db containment.fasta -parse_seqids -db_type nucl
$ blastn -db containment.fasta -query scaffolds.fasta -max_hsps 1 -outfmt 6 -out result
# blast 结果的相似性筛选,小于90认为与污染物种不同。将过滤的片段长度求和,判断过滤的片段是否与物种基因组大小一致。如果接近,那么即使有部分片段遗漏,但是大部分基因组数据已经保留。
$ awk '{ if ($3 < 90) print $1 }' result > filter_nodes
$ awk -F'_' 'BEGIN {len=0} {len+=$4} END {print len}' filter_nodes
# 进一步用目的物种的参考基因组进行blast,以确保没有其他物种污染。
$ makeblastdb -db reference.fasta -parse_seqids -db_type nucl
$ blastn -db reference.fasta -query assembly.fasta -max_hsps 1 -outfmt 6
用 Mapping 目的基因组来筛选 reads 再进行拼接
抓取目标nodes
# 保存代码到 get_nodes.py 文件中,运行 python get_nodes.py
from Bio import SeqIO
input_file = 'scaffolds.fasta'
filter_file = 'filter_nodes'
output_file = 'assembly.fasta'
wanted = set(line.rstrip("\n").split(None,1)[0] for line in open(filter_file))
print "Found %i unique identifiers in %s" % (len(wanted), filter_file)
records = (r for r in SeqIO.parse(input_file, "fasta") if r.id in wanted)
count = SeqIO.write(records, output_file, "fasta")
print "Saved %i records from %s to %s" % (count, input_file, output_file)
if count < len(wanted):
print "Warning %i IDs not found in %s" % (len(wanted)-count, input_file)